Projects
Molecular mechanisms of dwarfism and organ allometry in Atlantic salmon
Species radiations can be driven by changes in body proportions (allometric changes), that is, natural selection acting on size and not in shape. Discovering the genetic and developmental basis of allometry has both fundamental importance for understanding the emergence of biological diversity, as well as in practical terms such as in understanding cancers. Only a few allometry mechanisms are known to explain adaptive differences in organ size within a vertebrate species. Salmon show a fascinating adaptation of organ allometry associated with a trade-off in life history strategies; dwarf landlocked ecotypes prioritize egg size over egg number, and in consequence have ovaries that are 50% smaller compared to their sea-migrating congeners. The decreased ovary size in dwarf salmon provides a unique model to study how allometry emerges within a single species and interplays with life history strategy. My work in collaboration with Prof Dylan Fraser at Concordia University is dissecting the genotype-phenotype-fitness map of ovary allometry from the molecular to the species level with an integrative approach combining cellular genomics, gene editing, and a powerful quantitative genetics framework.

Molecular mechanisms of age at maturity variation in Atlantic salmon
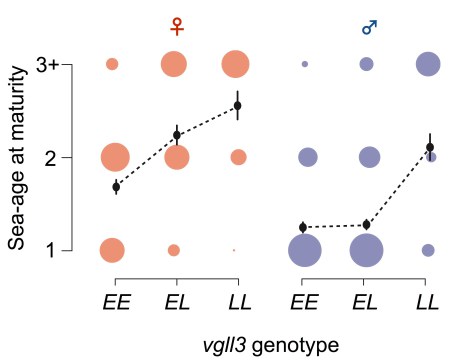
Trade-offs between life-history traits such as growth and reproduction are common in nature. One example of such evolutionary trade-off is seen in Atlantic salmon, which may grow to impressive size and produce thousands of eggs. Growing big in salmon is related to the number of years it spends at sea feeding. Feeding for a long period means potential for more offspring, but with the cost of increased risk of dying before reproducing. In Atlantic salmon this trade-off seems to be different for males and females - females tend to be bigger (and older) than males by the time the fish returns to its native river to spawn.
The genetic basis of age at maturity (when salmon return to spawn) has been mapped to a region of chromosome 25 containing the gene vgll3 (Barson, Aykanat et al. Nature 2015). Two alleles at vgll3 (E for early maturity and L for late) explain nearly 40% of variation in age at maturity. vgll3 is a transcription co-factor that is involved in fat tissue differentiation in fish and mammals, but how it could control for age at maturity is not known. During my post-doc at the Primmer lab, I studied the molecular mechanisms that translate vgll3 genotype variation into differences in age at maturity.
Genetic basis of gene expression divergence in threespine sticklebacks
Variation in gene regulatory mechanisms play an important role in producing phenotypic variation that natural selection can act upon. Regulatory evolution seems to be tightly linked to both adaptive divergence of lineages as well as divergence of established species. We have a poor understanding of regulatory evolution during the very early stages of incipient speciation where adaptation-with-gene-flow still dominates, as it does in sticklebacks. These fish are a great model to study regulatory evolution at short divergence timescales as many of the freshwater environments sticklebacks have colonised were formed after the last glaciation only ~10 000 years ago, and the repeated nature of freshwater adaptation provides a system where we can study regulatory changes that have occurred in parallel in independent bouts of adaptation.
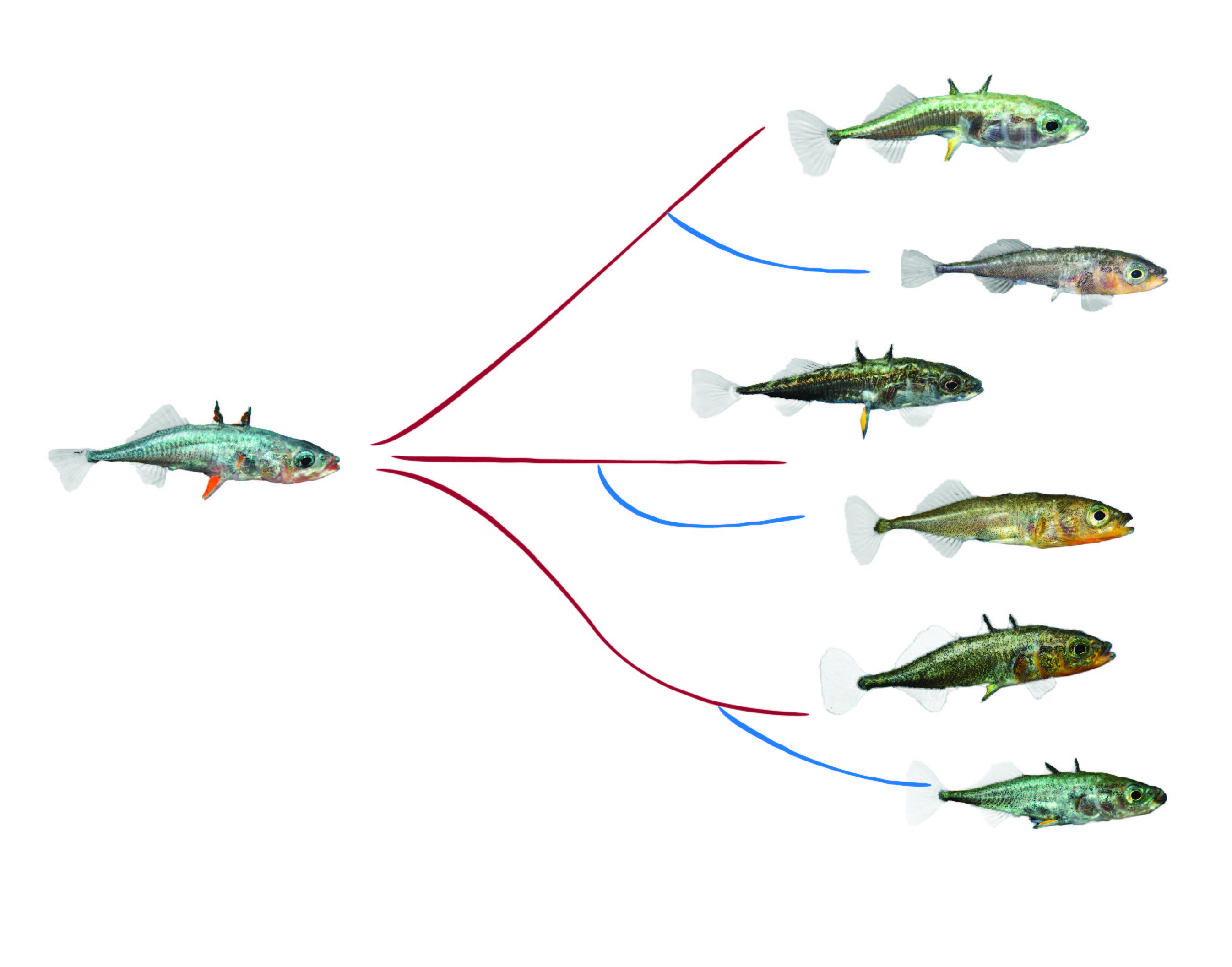 Stickleback freshwater forms have evolved from marine ancestors in parallel
Stickleback freshwater forms have evolved from marine ancestors in parallel
During my post-doct at the Jones lab (FML/MPI Tuebingen), I characterised gene regulatory divergence between freshwater and marine sticklebacks in order to better understand the contribution of different regulatory mechanisms to adaptation and incipient speciation. To reach this goal I used whole-genome and mRNA sequencing combined with bioinformatics and statistical analyses to explore the ways in which gene expression divergence in the gill tissue is linked parallel freshwater adaptation.
eQTL architecture and expression evolution in conifers
Understanding the genetic basis of variation in phenotypes is a central goal in genetics, and essential for deciphering how genetic variation facilitates evolution. Current approaches allow the genetic tracking of complex genomic phenotypes in only a few species, limiting the ecological and evolutionary contexts where the effects of genetic variation can be studied.
During my PhD I developed an approach to study the genetic basis of gene expression variation that can be applied to conifer trees. Conifers are foundation species of boreal forests that are increasingly threatened by effects of climate change such as extreme weather and insect outbreaks. It is therefore timely that we develop tools and approaches with which we can study microevolutionary processes in conifer trees and assess the adaptive potential of populations.
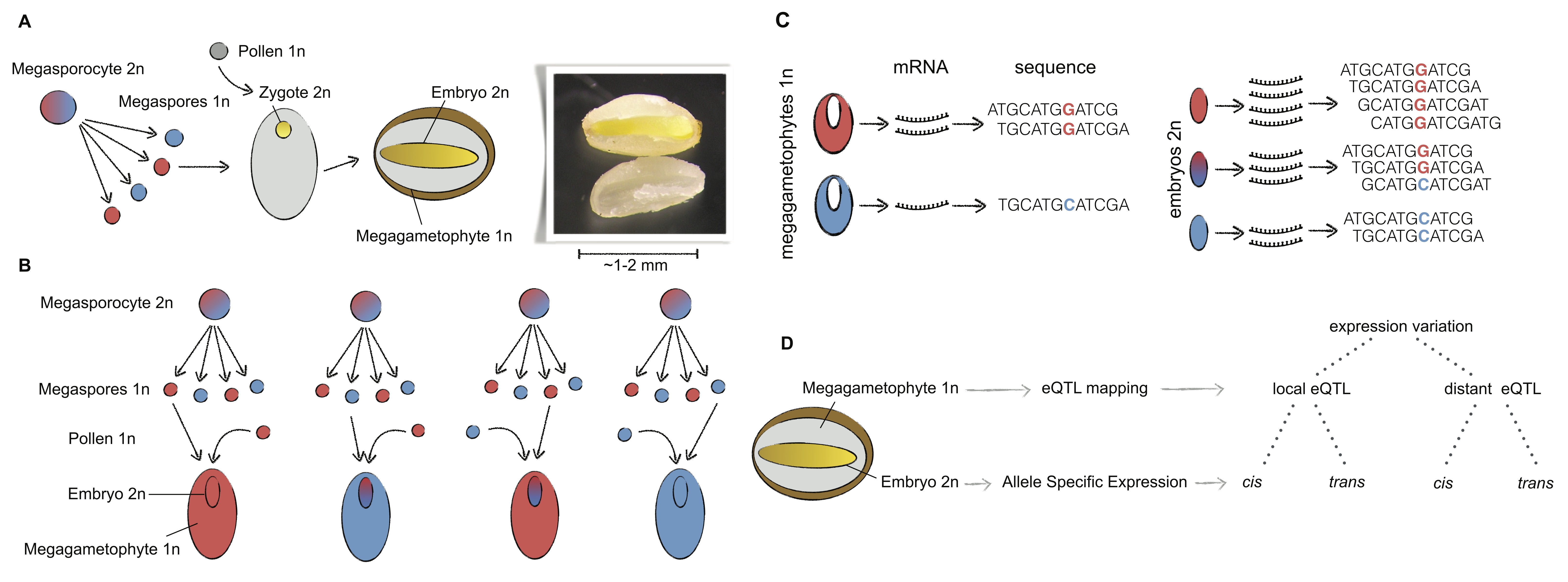 Conifer megagametophyte-embryo system to map eQTL
Conifer megagametophyte-embryo system to map eQTL
We developed an approach to map eQTLs based on analyzing gene expression in the haploid and diploid meiotic tissues of conifer seeds (see Papers). These tissues are conserved throughout the gymnosperms and therefore the approach opens the door for the genetic analysis of expression variation in a group of ~600 plant species.
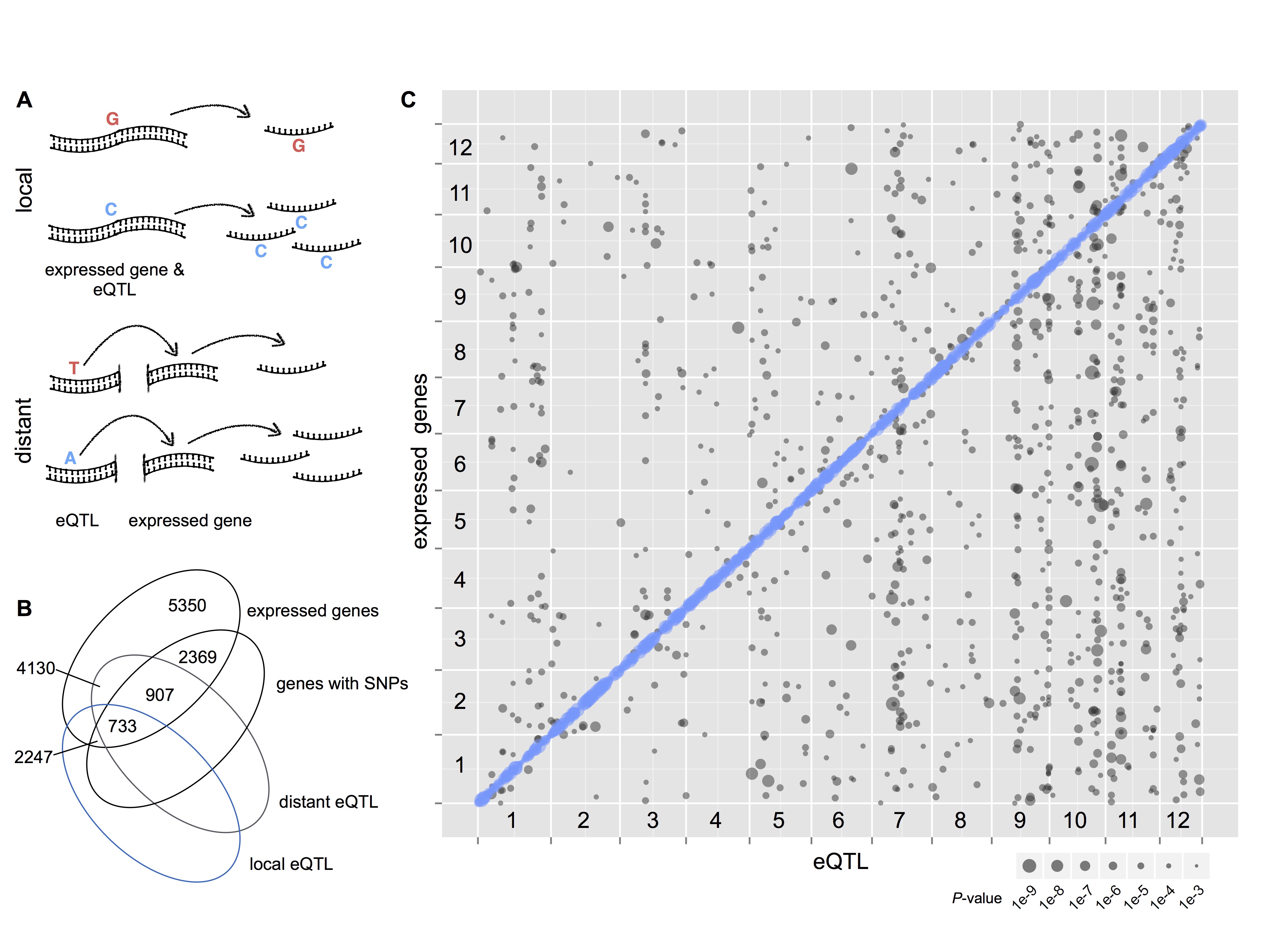 Spruce eQTL map built from haploid megagametophytes with RNAseq
Spruce eQTL map built from haploid megagametophytes with RNAseq
We used our approach to study the relationship between eQTL location and allele-specificity. An advantage of the approach is that it allows for a comprehensive characterisation of the genomic positions of mutations influencing the expression of genes and whether they were cis-acting (mutations in putative non-coding regulatory elements) or trans-acting (mutations in putative regulatory molecules such as transcription factors of regulatory RNA).
Our results point towards a number of interesting biological features of transcriptional variation in spruce. For example, we discovered genes that seemed to regulate their own transcription. We also discovered that co-evolution of different regulatory factors was often associated with a genomic architecture where the co-evolved factors were in close genetic linkage. We think that such an architecture might be favoured by natural selection as recombination between co-evolved regulatory factors has been shown to lead to extreme phenotypes - which is a form of genetic load. We observed that, overall, expression variation was associated with genes involved in stress-response, which may be a good sign for evolutionary potential in genes important for adapting to environmental changes.